Medical Device Risk Analysis
Risk analysis isn't a compliance check - it's a continuous engineering effort that shapes your device's safety and effectiveness from day one.
In medical device development, risk analysis is not a compliance check completed before a product launch. It is a continuous effort, an engineering tool that directly shapes the device and its safety and effectiveness. For quality engineers, design engineers, and project managers, establishing a bulletproof risk architecture is essential to protect patients, pass rigorous regulatory audits, and ensure successful product clearances.
True risk analysis is at the core of good medical device engineering. It bridges engineering creativity with maximizing patient safety and the harsh reality of regulatory requirements. It will help you develop the safest, most effective device possible. It directly shapes your hardware architecture, your user interface, and your software loops from day one.
The Regulatory Framework: ISO 14971
Global regulatory bodies demand a structured, lifecycle-focused approach to medical device risk management, and that is primarily defined by ISO 14971. The standard demands a continuous lifecycle process. You must identify hazards, estimate and evaluate risks, implement controls, demonstrate they are effective and monitor how those controls perform in the real world.
FDA Guidance and Human Factors Engineering
The U.S. Food and Drug Administration (FDA) aligns directly with ISO 14971 but places an intense, distinct emphasis on use-safety. The FDA Human Factors Guidance requires manufacturers to optimize the user interface to eliminate or minimize use-related risks.
Whereas most risk analysis methods consider the severity of the harm as well as the probability of it happening, the FDA's Use-Related Risk Analysis (URRA) Guidance establishes that only the potential severity should be considered when determining critical tasks.
Core Methodologies of Risk Analysis
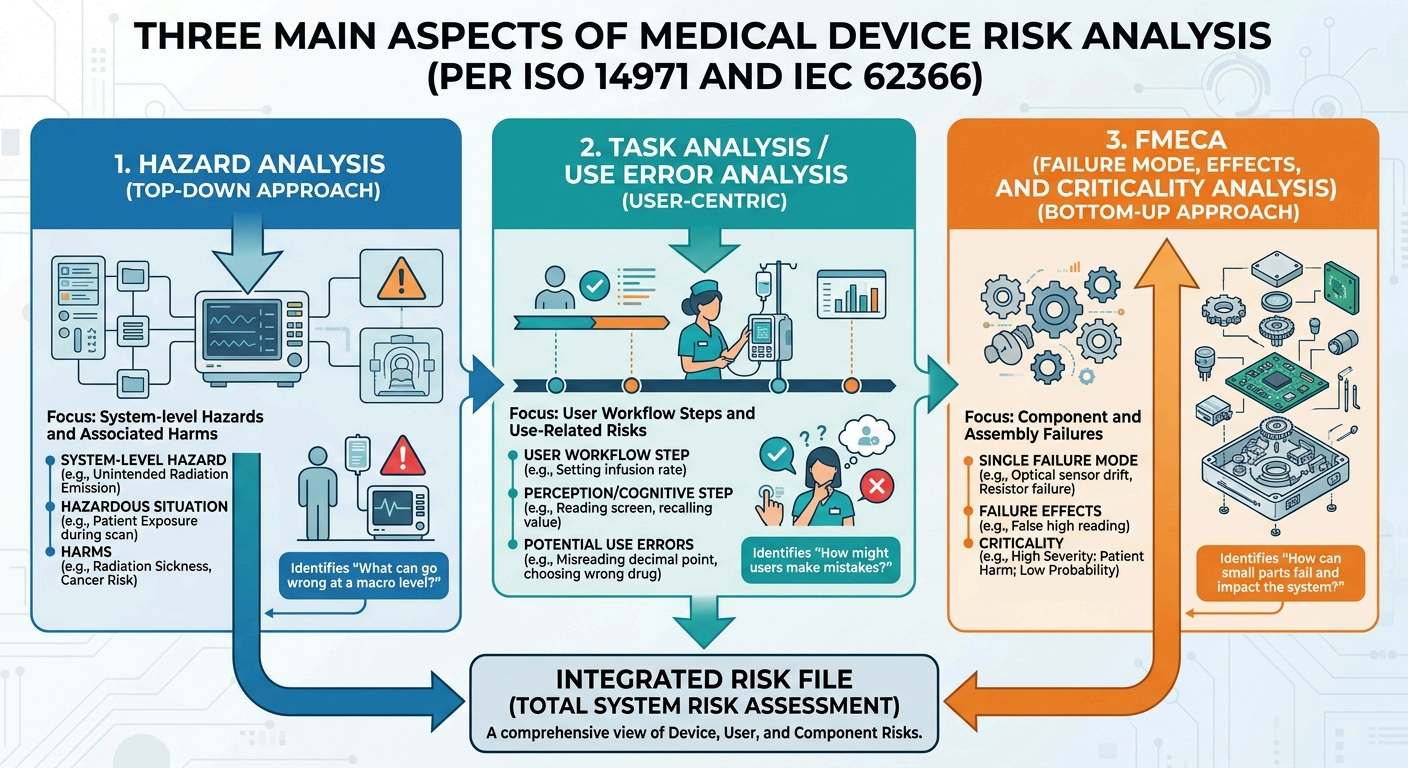
A comprehensive risk management file (RMF) relies on three core analytical pillars: Hazard Analysis, Task Analysis / Use-Related Risk Analysis (URRA), and Failure Modes, Effects, and Criticality Analysis (FMECA). Each addresses a distinct dimension of device failure.
1. Hazard Analysis
- What It Is: A top-down, system-level evaluation of a device's inherent characteristics under normal and fault conditions.
- The Goal & Benefits: To systematically uncover all potential sources of harm.
- What is Involved: Map out the relationship between a Hazard, a Foreseeable Sequence of Events, a Hazardous Situation, and the resulting Harm.
- Example:
- Hazard: High voltage electrical energy.
- Sequence of Events: Device enclosure cracks > fluid leaks into the housing > user touches the wet exterior during operation.
- Hazardous Situation: User is exposed to live high-voltage current.
- Harm: Severe electrical shock or cardiac arrest.
2. Task Analysis and Use-Related Risk Analysis (URRA)
- What It Is: A user-centric analysis focused on the steps required to prepare, operate, maintain, and decommission a medical device.
- The Goal & Benefits: To identify critical tasks — user interactions where an error could result in significant patient or user harm. It ensures the physical user interface, labels, and training effectively prevent use errors.
- What is Involved: Decompose the user workflow step-by-step. For each step, they analyze:
- What is the user's perception, cognition, and action?
- What use error could occur (e.g., misreading a screen, skipping a step, reversing a component)?
- What is the clinical consequence of that error?
- What risk controls to implement to mitigate it?
3. Failure Modes, Effects, and Criticality Analysis (FMECA)
- What It Is: A bottom-up, highly detailed technical approach applied to design architecture and manufacturing processes.
- The Goal & Benefits: To evaluate component-level or process-level vulnerabilities, assessing their local effects on the system and overall criticality to the patient.
- What is Involved: Break down physical components (e.g., device components, software subroutines, electronic hardware). Define exactly how a component could fail, the probability of that failure occurring and the final clinical severity.
Why Legacy Methods (Spreadsheets) Struggle in Risk Analysis
Most engineering departments historically relied on static, disconnected spreadsheets and other documentation for risk files. In modern medical device design, this siloed tracking causes major problems:
- The Risk-Traceability Disconnect: ISO 14971 explicitly requires all risk mitigations to be verified. This requires tracing your risk controls across requirement documents and verification protocols and tests. With such complexity, it is easy to miss or break a link as things develop and move around.
- Orphaned Mitigations: When design specifications change late in a project, manual spreadsheets make it difficult to find what gaps remain.
- Unification: When teams of people are working across multiple documents, it's difficult to stay unified on terminology, harms, hazards, definitions of probability and severity, etc.
Don't Make It So Easy for Them

Auditors love to search for inconsistency. You have 3 different documents, saying 3 different things, and you're on the fast-track to an observation or finding. It doesn't have to be that way.
Unified Requirement and Risk Analysis Development and Tracing
If you want to get through the design control process without drowning in a messy web of documentation, follow these three rules:
- Stop separating risk from requirements: Write your user needs, design requirements, and your hazard analysis simultaneously. Easily track your risk mitigations across multiple documents. From every risk to requirement, protocol to test, the path needs to be clear. Purpose-built applications can unify your Hazard Analysis, URRA, and FMECA data with your product requirements and testing. These applications let you see the big picture and dive into the details in the same place.
- Ditch manual tracking: Stop risking your regulatory submission with a mess of questionably connected documents. Modern requirement, risk analysis and traceability tools are more effective and affordable than ever. Let them take some pain out of the documentation so you can focus on what you actually love, engineering great products.
- Know where you stand: The last thing you want in developing your medical device is to be wandering around, lost in the woods. Give yourself the gift of instantly knowing where you are, how much further you have to go and where your gaps are. Internally auditing your risk analysis documentation and traceability can take days if not weeks when spread across multiple spreadsheets and documents. Risk Analysis and Requirements traceability software can do it in an instant. Know at any time if you're fully compliant or where to focus your attention. Show that to your auditor and give them the confidence to just move on to the next topic.
For a long time, startups and lean engineering teams were priced out of these sorts of tools. The enterprise software companies went after the big whales of medical device and unless you had deep pockets, you were forced back into spreadsheet hell.
Luckily, those days are over. Modern, purpose-driven risk and requirement traceability applications are highly accessible. You can develop and link your Hazard Analysis, FMECA rows, and Requirements Traceability into a single source of truth for a fraction of historical legacy costs.